所有计算课程已经不再与研之成理合作,购买课程请联系微信:13521890527(小计算)
中级班培训为期4天(线下,每天8小时),涵盖了1000多页PPT,几十个实战算例,计算案例操作视频,现场提供免费的超算上机练习。经过刘锦程博士一年多精心备课,此次培训通过深入表面计算领域最前沿,解密计算TOP期刊!
建议:中级班课程要求学员曾上过初级班,或者有至少一年以上的第一性原理计算经验!(初学者请先参加初级班,否则可能中级班完全跟不上!)
理论计算的文章要怎样发到JACS及以上的期刊?除了好的Idea,在技术上有四大法宝,中级班就是围绕这四个主题展开的。
- 热力学分析
- 动力学分析
- 电子结构分析
- 从头算分子动力学
- 高通量计算+机器学习
1. 表面催化反应热力学和动力学
表面与表面能
原子堆积方式,FCC、HCP、BCC,Miller指数,高miller指数晶面与台阶面,表面弛豫与重构,对称和非对称的slab模型,对称和非对称表面的表面能计算,金属表面能和功函数据库,原子坐标固定,纳米颗粒的结构和表面能关系,Wulff construction计算,纳米催化中的晶面效应。
表面吸附
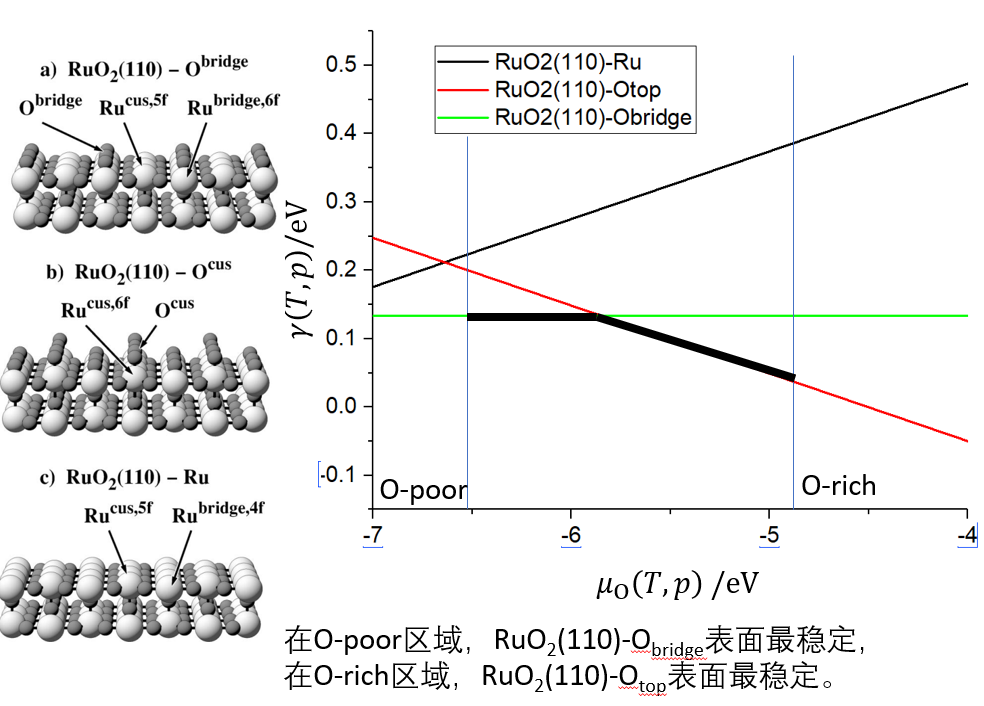
吸附类型(化学吸附、物理吸附、解离吸附),吸附能计算,吸附位点,吸附计算注意事项,吸附物种间的相互作用,覆盖度,平均吸附能,最后一分子的吸附能。化学平衡,表面吸附平衡,Langmuir吸附等温式与前提假设,熵变对吸附的影响,覆盖度和温度/压力/吸附能关系计算,多组分气体吸附平衡,催化剂中毒微观原理,表面化学势的计算与表面热稳定性计算,催化剂中毒常见案例。表面催化计算研究思路。
过渡态搜索
过渡态判据,无过渡态反应类型,寻找过渡态方法综述,chain-of-states搜索方法和原理,传统PEB方法(plain
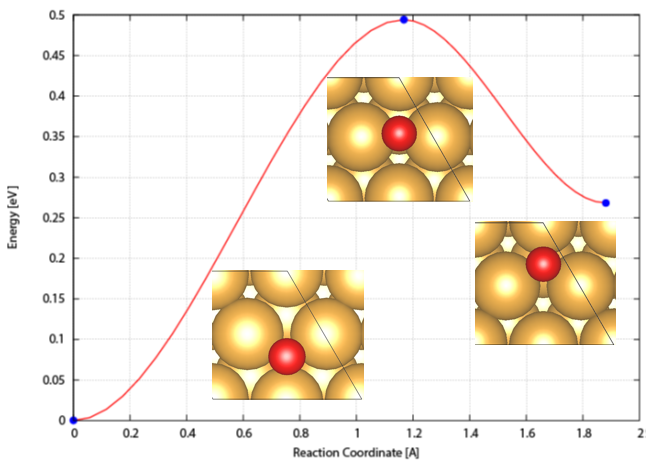
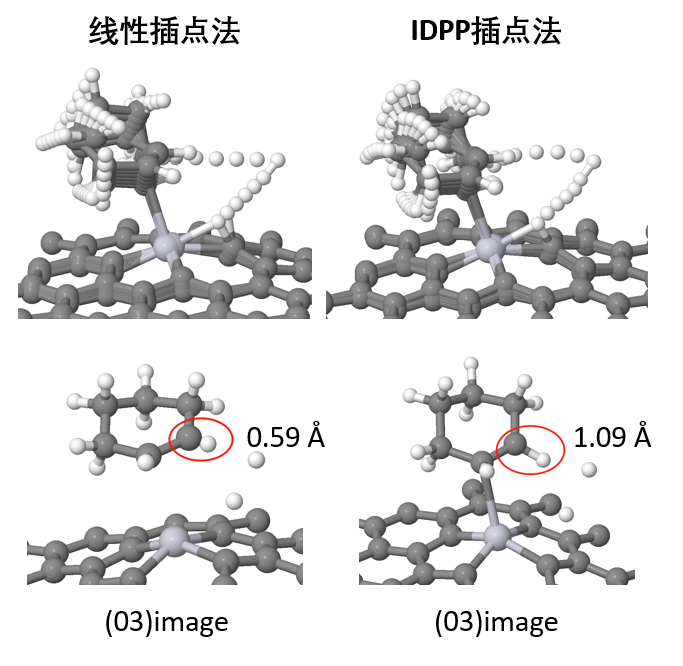
elastic band)方法遇到的问题,NEB中nudge过程原理,Climbing Image原理,判断插点个数,插点方法,nebmovie.pl生成movie判断插点合理性,CINEB的INCAR关键词和计算方法,VTST优化算法解析,nebresults.pl,nebbarrier.pl, nebspline.pl, nebef.pl等VTST脚本,neb.dat、spline.dat、exts.dat、mep.eps文件自动判断NEB路径上的极值点,Dimer方法原理,选择Dimer,平移Dimer过程,Dimer发展历史,Dimer的INCAR关键词和计算方法,MODECAR生成,DIMCAR、CENTCAR和NEWMODECAR解读,生成DIMER方向movie预判过渡态计算是否成功,VTST过渡态计算收敛判断,过渡态搜索常见问题和解决方案。扩散系数与离子电导计算。NEB+Dimer高效搜索过渡态,neb2dim.pl。NEB计算线性插点的问题,IDPP插点原理,使用pymatgen-diffusion和pymatgen做IDPP非线型插点。
频率计算
Hessian矩阵,力常数矩阵对角化,孤立和吸附分子振动区别,零点振动能,有限位移和DFPT方法频率计算INCAR,Jmol观察振动,振动数据提取和分析。
热力学数据计算
系综配分函数,分子配分函数,热力学量与配分函数的关系,内能、亥姆霍兹自由能、焓、吉布斯自由能、熵、等容热容、等压热容,理想气体假设,能量成分的拆分,平动、转动、振动、电子跃迁的配分函数,和其对各种热力学量的贡献计算,振动频率、温度和熵的关系,0K热力学数据校正计算,实际温度热力学数据校正计算,VASPKIT计算吸附分子热力学量,吸附分子和孤立分子的热力学量计算区别。气体分子的自由能校正,查实验数据热力学表JANAF-NIST,使用Gaussian程序做频率计算得到热力学数据,VASPKIT计算孤立分子热力学量。
化学反应速率常数计算
基元反应,单分子多分子基元反应速率方程,稳态平衡和速率方程关系,反应速率常数计算,Arrhenius equation,eyring equation推导,指前因子计算,表观活化能拟合,过渡态理论,准平衡态假设,过渡态络合物,透热系数,Recrossing问题,判断基元反应是否容易发生,Eyring过渡态理论的局限性,ΔGa/温度/反应速率常数k之间的关系,半衰期,谐振过渡态理论HTST公式,吸附和脱附步骤反应速率,Knudsen-Langmuir equation,隧穿效应,Wigner方法计算透热系数。
催化反应机理
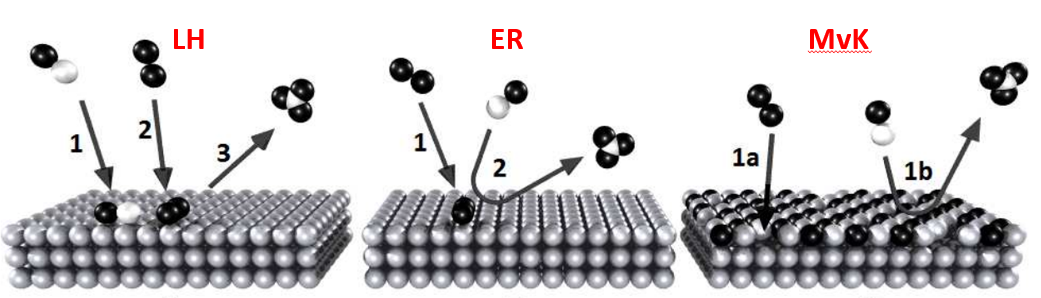
Langmuir-Hinshelwood (LH) mechanism,Eley-Rideal (ER) mechanism,Mars-Van Krevelen (MvK) mechanism,及其速率方程组。LH实例合成氨反应,ER实例Volmer-Heyrovsky HER,MvK实例低温CO氧化反应。催化循环和总包反应能量。
微观动力学模拟简介
TOF(Turnover frequency),TON(Turnover number),微观动力学模拟,稳态近似,平均场近似,联立和求解微分方程组,CatMap的使用,CatMap 输入文件,形成能的计算,shomate_gas,绘制速率-温度/压力图,输出覆盖度图,提取自由能势能面,绘制吸附能火山曲线,定义linear scaling,拟合BEP关系线。
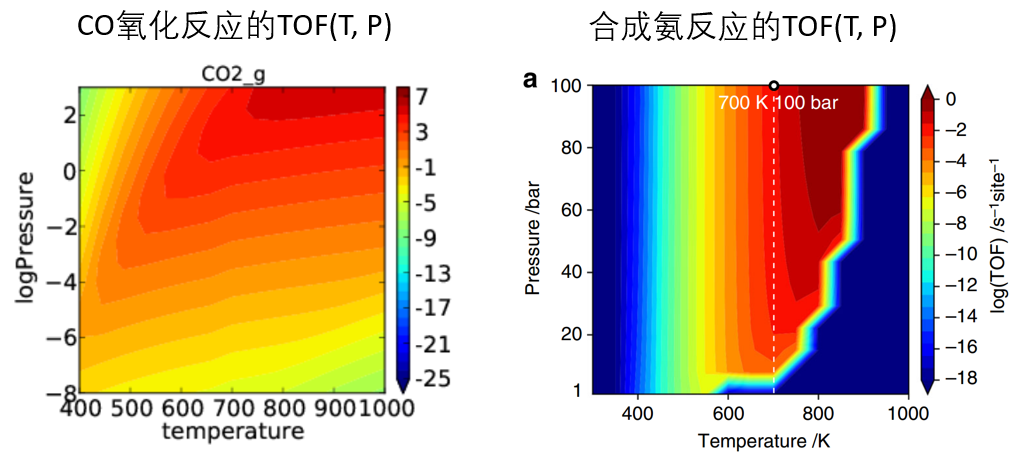
决速步和决速态理论
决速步,决速步理论推导出解析的TOF的表达式,温度/分压对TOF影响,k表象,E表象,决速态,TOF-determining transition state (TDTS),TOF-determining intermediate (TDI)。
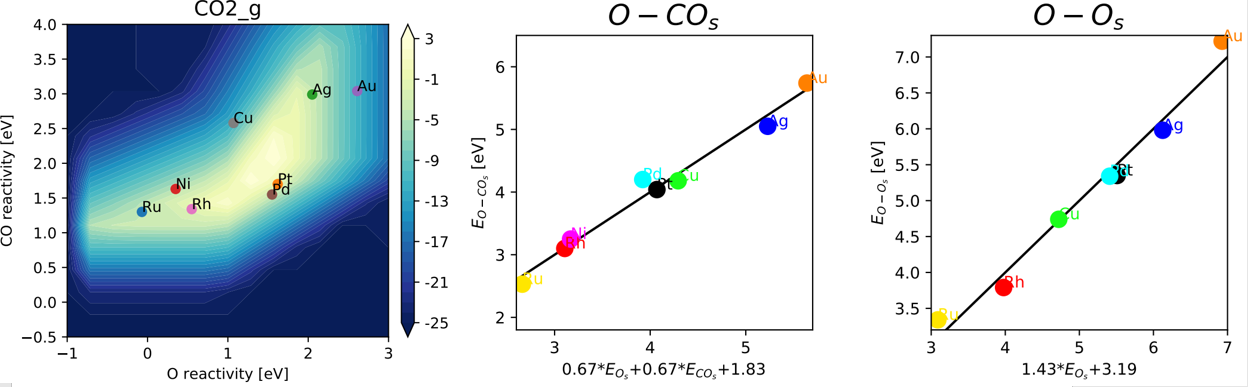
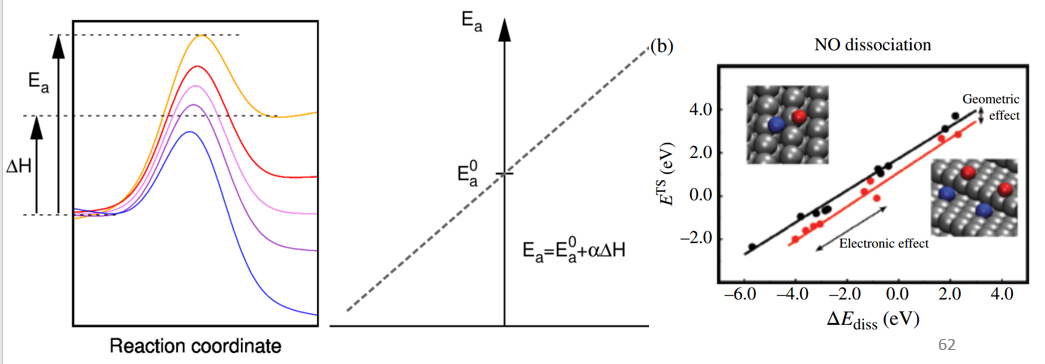
BEP和火山曲线
BEP关系,甲烷C-H活化/NO分解等催化反应中的BEP关系,BEP关系注意事项,截距和斜率规律。BEP关系最新拓展:配位数,d带中心,态密度,费米能级,电离能,功函等等的线性规律。
高通量计算与机器学习
Materials project数据库API使用和批量获取结构方法。Pymatgen简介,自动化表面切割,自动生成表面吸附结构,自动绘制能带结构,态密度图,稳定性相图。目前较为成熟机器学习软件简介:LASP,DP-GEN,SISSO,CGCNN,AiiDA等。
2. 电子结构分析
常见VASP第一性原理计算中的电子结构分析手段总述。
态密度
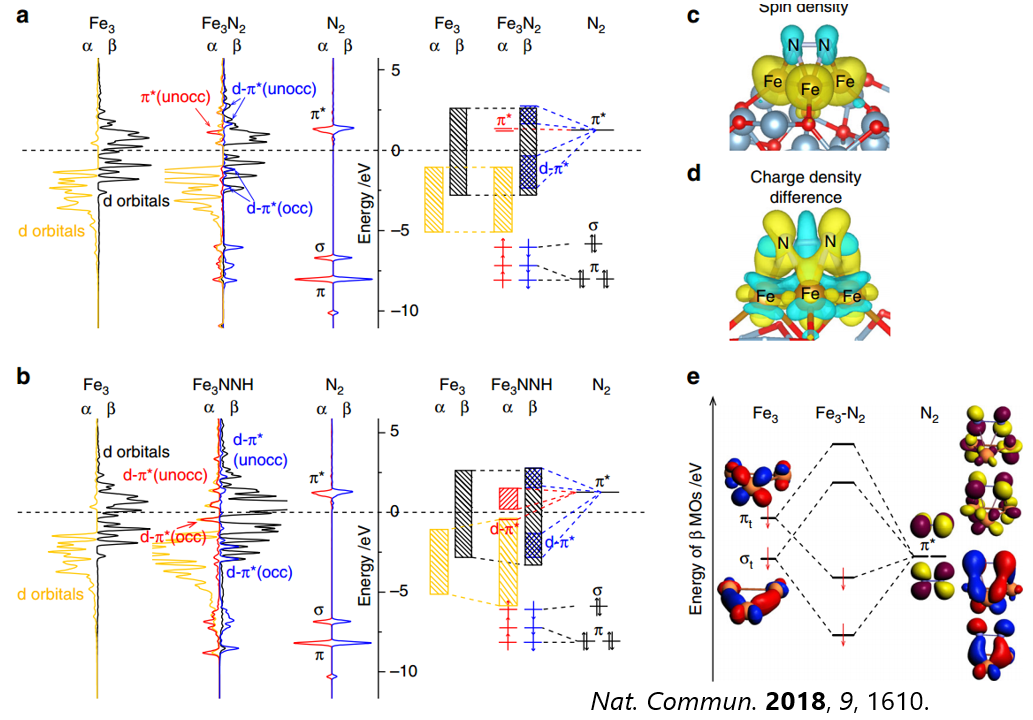
态密度和能带形成的基本理论,案例分析(单原子催化剂,表面离子缺陷态,表面态,d带中心,催化吸附的轨道相互作用模型),态密度计算方法,p4vasp和VASPKIT后处理分析,态密度的积分,pymatgen自动画态密度图,K点选取和ISMEAR对态密度计算影响,DOSCAR详解,过渡金属金属的d带中心计算和催化吸附的关系计算,N2分子和表面团簇催化剂的轨道相互作用模型分析。
轨道相互作用基础知识,高斯计算提取分子轨道,载体和吸附分子态密度作用分析,CO/Ni,N2/Fe3催化剂的轨道相互作用模型分析。
电荷密度和原子电荷
CHCGAR的格式,分解CHGCAR成tot和mag,VESTA作等值面图,二维切面图。VASPKIT处理电荷密度文件,电荷密度差分,吸附分子CO,N2吸附前后的电荷密度差,石墨烯插锂前后的电荷密度差;变形电荷密度;外场下的电荷密度差;平面平均的电荷密度差。Partial charge density计算方法流程,实空间波函数提取,绘制分子的分子轨道图,绘制VBM和CBM的电荷密度/波函数,绘制缺陷态/隙间态的电荷密度/波函数,研究激发电子的分布,STM计算和Partial Charge Density对应关系,STM“针尖偏压” 和 “恒定高度”模拟。Bader电荷,全电子电荷密度生成,AIM(atom-in-molecule)电荷划分方法,按照原子电荷着色的结构图。
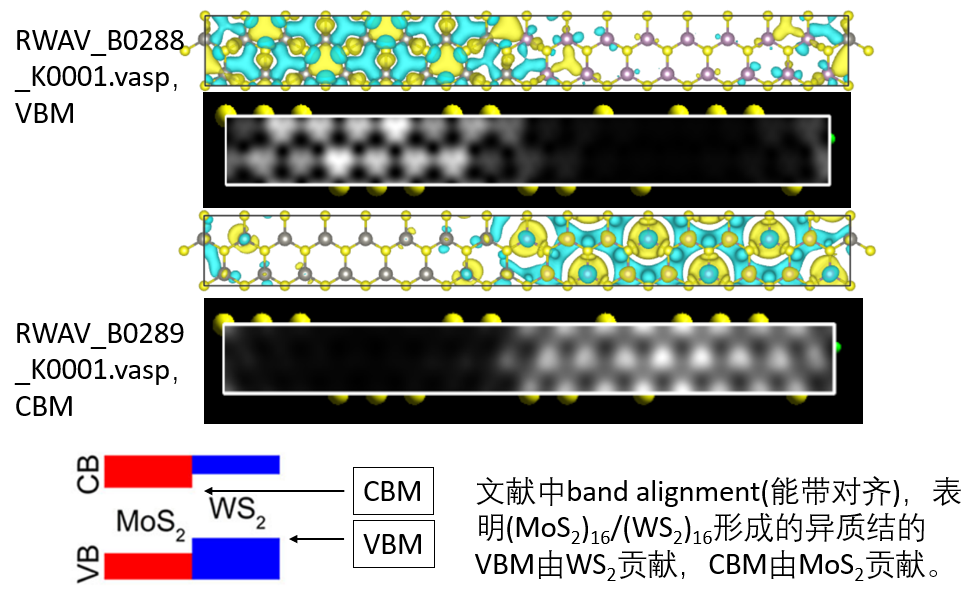
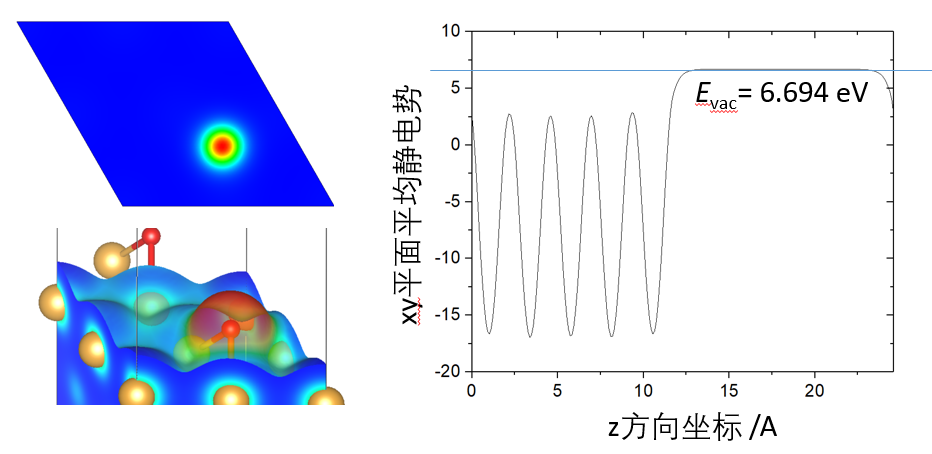
ELF、静电势和功函
功函数,分析异质结静电势,静电势分析表面负载团簇的电子流向,分子表面和载体表面静电势分布,受阻路易斯酸碱对分析。电子定域化函数(ELF),ELF研究表面催化反应过渡态、电子化合物、缺陷态、分子团簇、吸附分子。
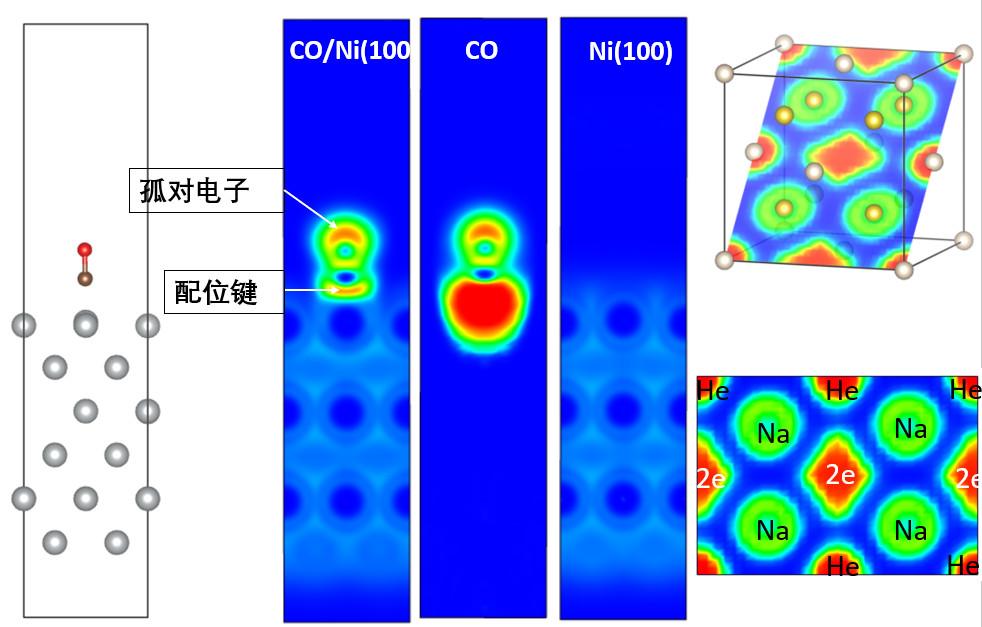
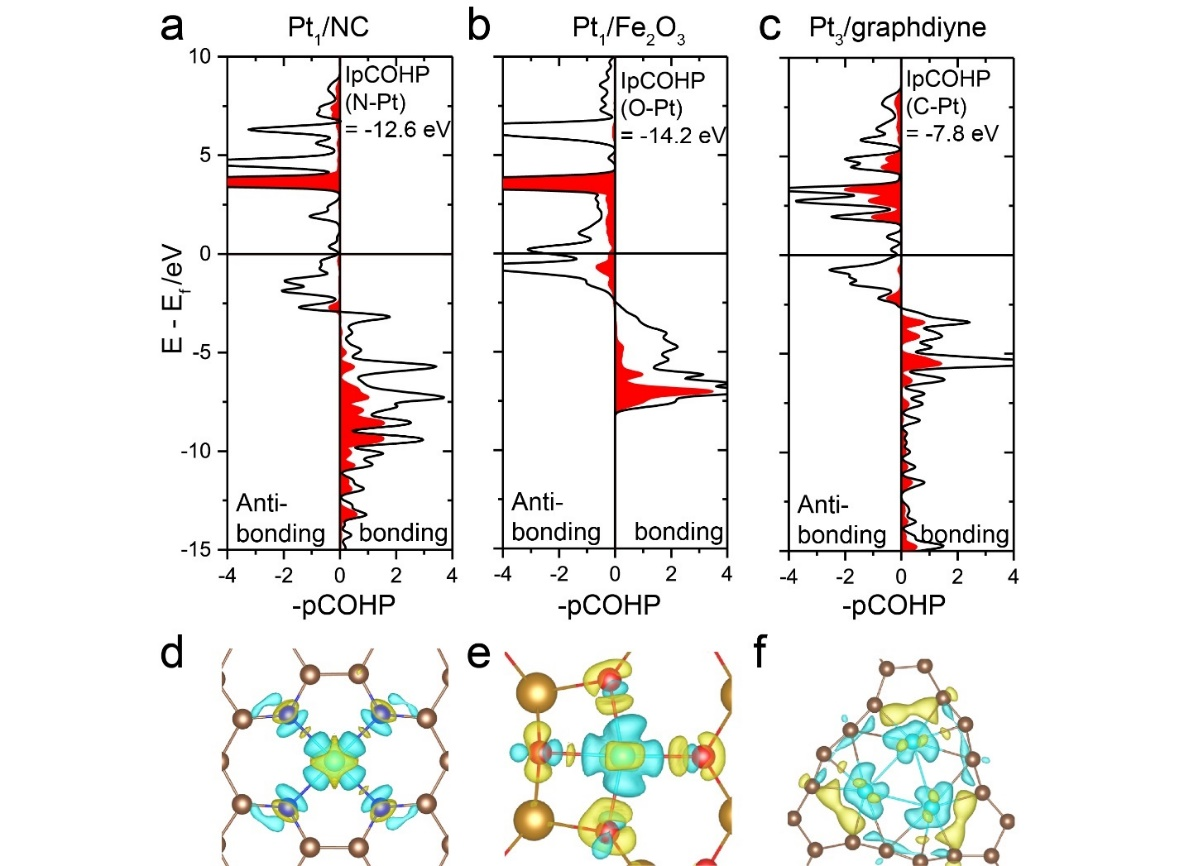
COOP、COHP
晶体轨道重叠布居COOP和晶体轨道哈密顿布居COHP基本原理,案例:COHP用于研究单原子Pt催化剂和CO的相互强弱,COHP用于NRR电催化反应分析,用于研究磁性影响的晶体相变,高压固体研究,镧硼新材料。Lobster计算COOP和COHP,Wxdragon使用,计算COHP的INCAR和注意事项,lobsterin输入文件,投影到基组的类型,投影到基组做包含的轨道,Spilling误差,指定原子对,指定距离范围,高斯展宽,打印Mulliken and Löwdin电荷,Lobster计算重启,输出轨道的实空间轨道(PAW或LCAO)数据。Lobster计算实例:金刚石,CO/Ni,孤立CO分子。
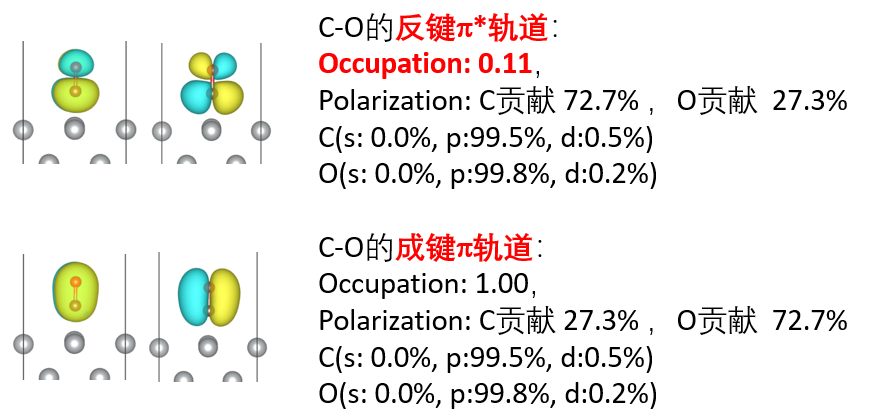
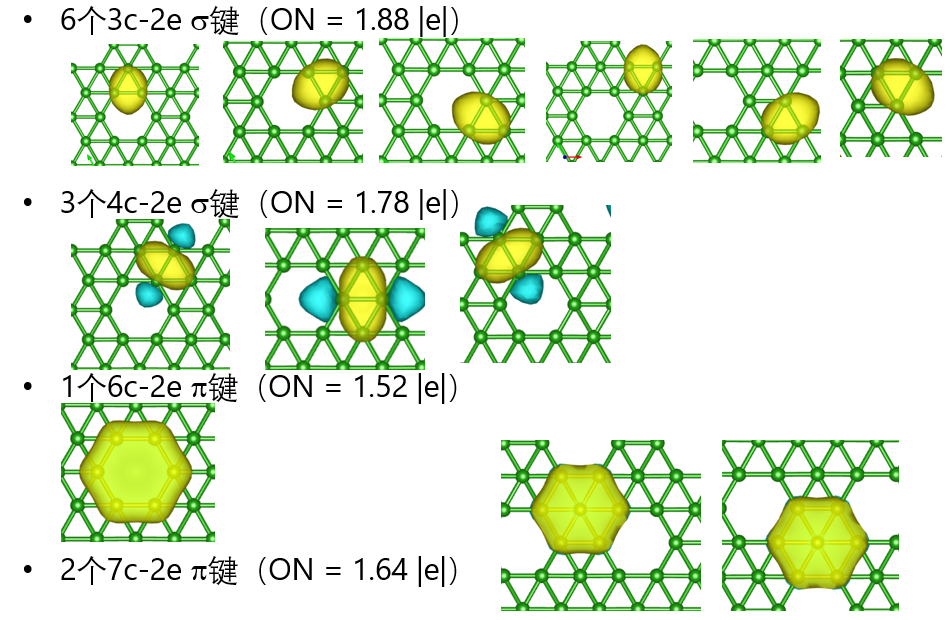
periodic NBO、ssAdNDP
电子定域化方法简介NBO和Periodic NBO 概述,对密度矩阵部分对角化+轨道权重的正交化,平面波基组到高斯基组的投影方法,NBO和Period NBO程序介绍,提取单中心,双中心的NBO。NAO(Natural atomic orbital)的轨道系数、NPA(Natural Population Analysis)电荷,Periodic NBO 程序编译,投影程序的编译projection.exe,basis.inp高斯基组文件准备,EMSL/斯图加特等主流基组库使用。实例:晶体Si的NBO计算,NBO轨道绘制和NBO计算结果分析。实例: CO/Ni。ssAdNDP,NBO方法的局限性,多中心化学键,AdNDP计算流程,ssAdNDP编译,ssAdNDP应用实例2D Borphene,2D B-Metal,Be5C2,Cu2Si,ssAdNDP的搜索技巧,原文还原:硼墨烯的多中心键研究。
3. CP2k与从头算分子动力学
CP2K编译:/tools/toolchain方法,预编译版本。
CP2K简介:CP2K的优缺点,CP2K经典文章介绍,
CP2K的使用:CP2K输入文件,cp2k.inp文件格式,CP2K计算原理和重要关键词详解,CP2K输出文件,CP2K基组,赝势,坐标读入,基组重叠误差,OT和Diag对比,结构优化和实例练习,Cu(111) + 46H2O 结构优化,建立固液界面模型,Au20/TiO2(110) 表面的优化。DFT+U,杂化泛函,磁性排列,范德华校正等参数的设置。
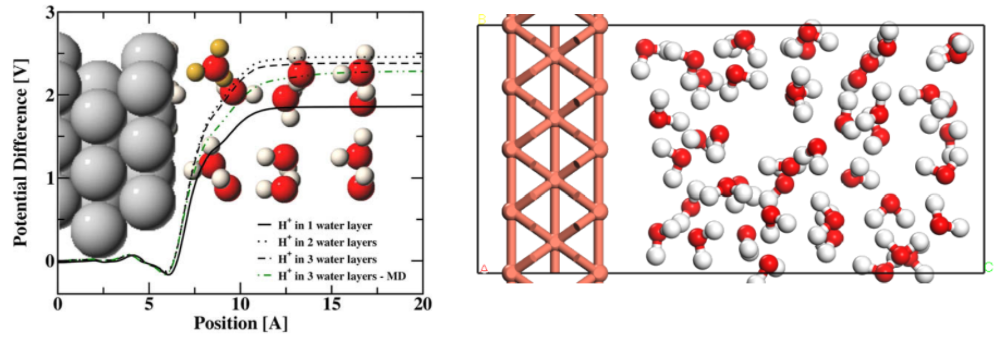
从头算分子动力学模拟:AIMD计算基本流程,相空间,系综,宏观量的计算,牛顿运动方程,速度形式的 Verlet 算法,AIMD模拟的关键参数,AIMD模拟时间步长的选择,AIMD模拟参数测试,AIMD的重复性问题,Energy drift问题,周期性边界条件,动能与温度,控温技术与热浴,速度调节法,Anderson热浴,Nosé–Hoover热浴,模拟退火,CP2K和VASP做AIMD的计算参数详解。轨迹文件的简化和可视化,VMD的简单使用,AIMD的后处理,键长键角跟踪,能量与温度涨落,径向分布函数,亥姆霍兹自由能路径计算,均方位移,扩散常数,速度自相关函数,红外光谱,固液界面平均静电势计算,引入电极电势对固液界面的影响。锂离子电池的动力学模拟。
自由度限制的AIMD:VASP和CP2K计算Metadynamics的方法。高斯势函数的参数技巧,数据后处理,自由能势能面,potential mean force,blue-moon/Slow-growth方法计算自由能势能面。
4. JDFTx与电催化计算
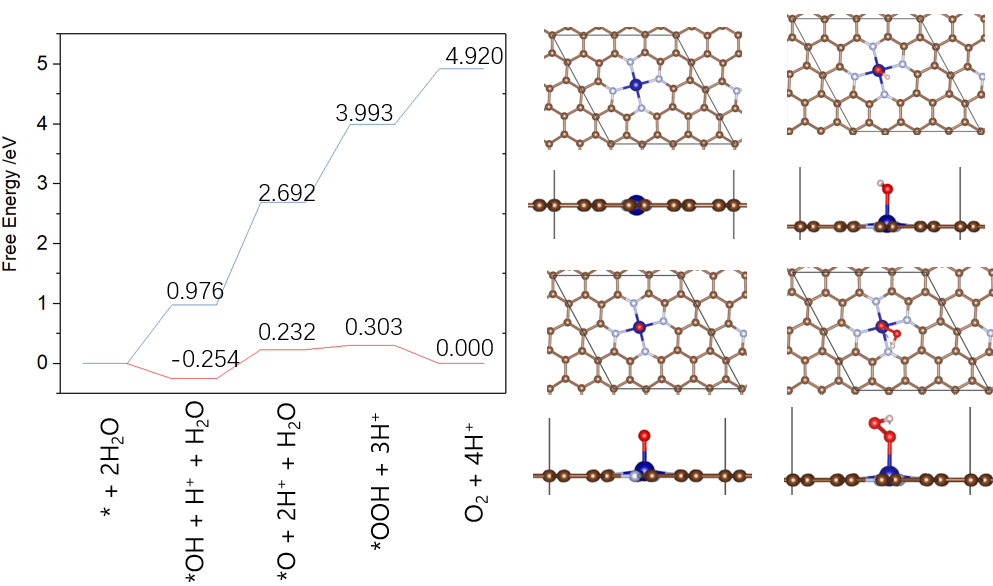
1.计算电催化基础:ORR、OER、HER、HOR反应机理,Norskov模型过电势计算,台阶图计算,电催化基元步骤自由能变计算,OER实例,M-N4/graphene,溶液的pH影响,NRR,CO2RR机理。
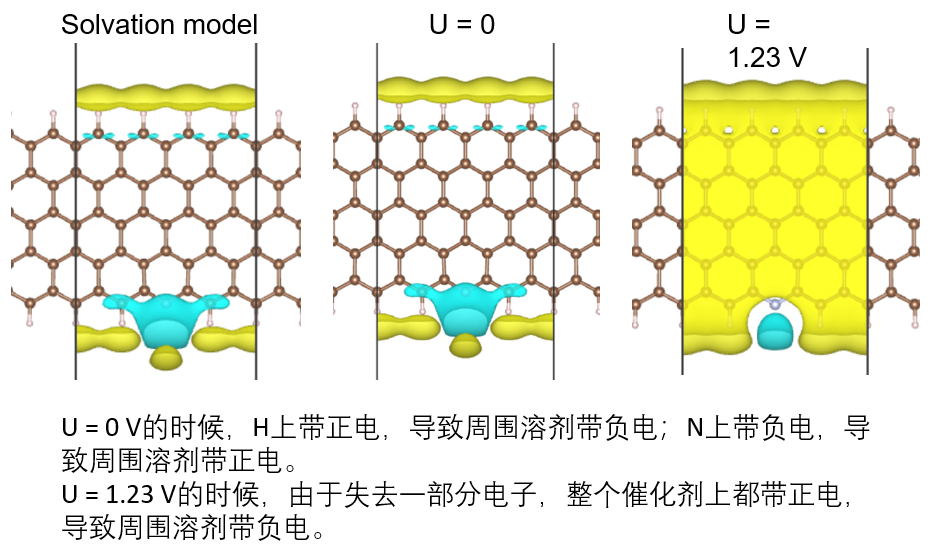
2. 进阶电催化计算:传统计算模型缺陷:“双电层”理论,Gouy-Chapman模型,零电荷电势potential of zero charge (PZC),显式溶剂模型流派和代表文献,显示溶剂模型流派和代表文献,连续介质模型,VASPsol使用方法和实例练习,JDFTx编译安装,JDFTx学习方法,JDFTx 输入输出文件结构,单点能计算.in文件,几何优化.in文件,频率计算.in 文件,溶剂化模型,bound charge,表面模型计算+溶剂化+电极电势计算,charge-neutral method vs constant-potential method,VASP配合JDFTx精确计算电催化模型的使用方法。溶剂和电极电势对二维材料电催化的影响。
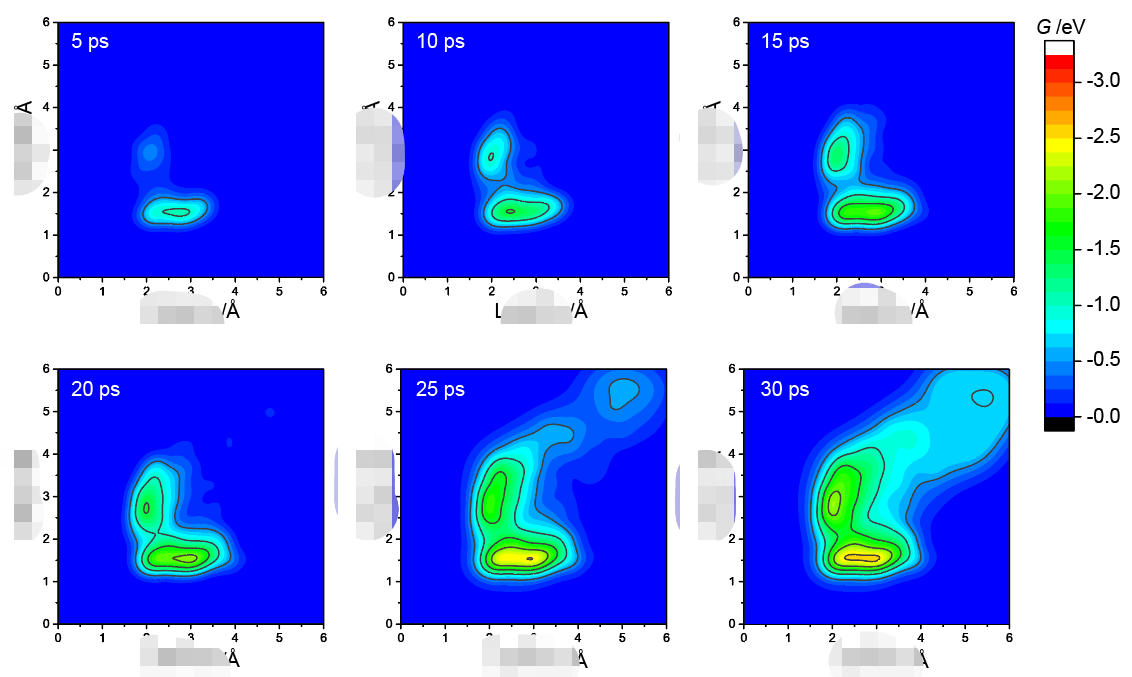
3. 显示溶剂模型+AIMD处理电催化界面方法(AIMD部分讲解)
转载请注明来源,欢迎对文章中的引用来源进行考证,欢迎指出任何有错误或不够清晰的表达。